
XOSPATA® (gilteritinib) Approved by U.S. FDA for Adult Patients with Relapsed/Refractory Acute Myeloid Leukemia (AML) with a FLT3 Mutation
– FLT3 mutation-positive AML is a life-threatening disease1 with significant unmet medical needs –
– XOSPATA is the first and only FLT3 inhibitor approved by the FDA for patients with relapsed or refractory AML with a FLT3 mutation –
– Approval of XOSPATA marks Astellas' U.S. entry into the treatment of blood cancers –
TOKYO – November 28, 2018 – Astellas Pharma Inc. (TSE: 4503, President and CEO: Kenji Yasukawa, Ph.D. “Astellas”) today announced that the U.S. Food and Drug Administration (FDA) approved XOSPATA® (generic name: gilteritinib) for the treatment of adult patients who have relapsed or refractory (resistant to treatment) Acute Myeloid Leukemia (AML) with a FLT3 mutation as detected by an FDA-approved test.2 XOSPATA is an oral therapy and the first and only FLT3-targeting therapy to be approved by the FDA for this population.
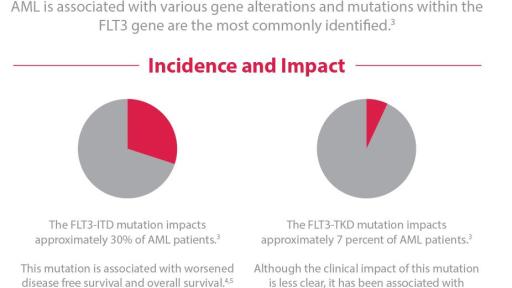
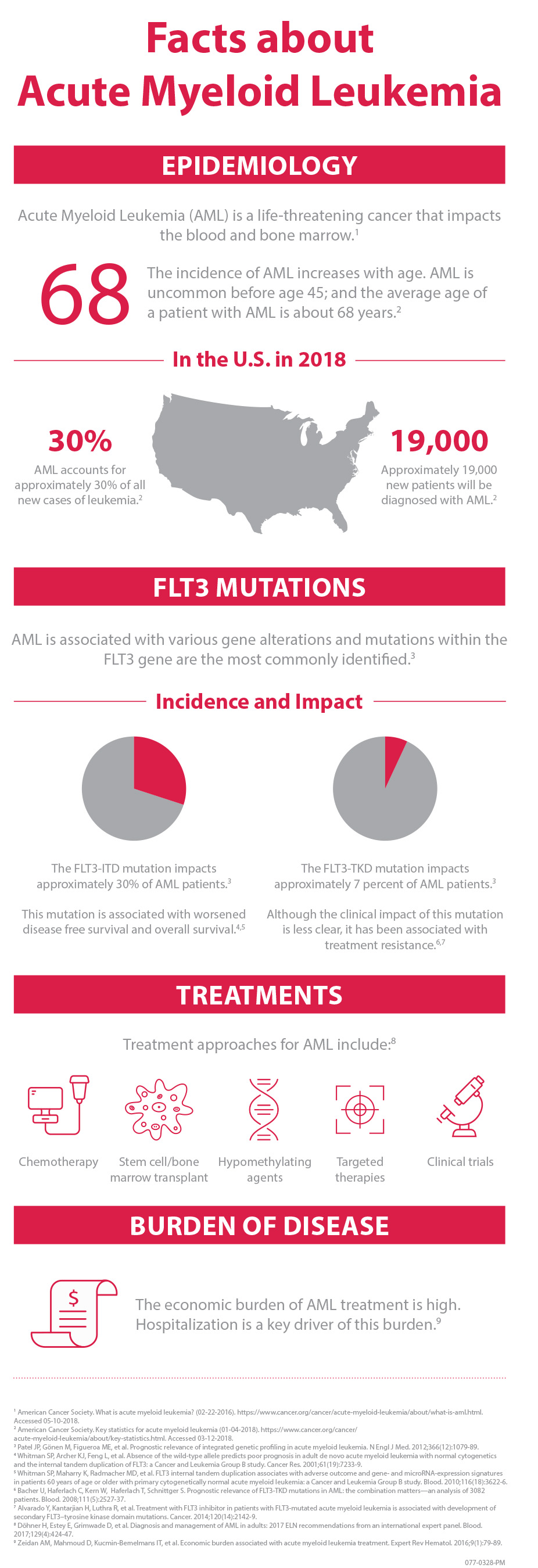
The American Cancer Society estimates that in 2018, approximately 19,000 people will be diagnosed with AML in the U.S.3 AML has been associated with various genetic mutations. XOSPATA has demonstrated inhibitory activity against two different mutations, FLT3 internal tandem duplication (ITD) and FLT3 tyrosine kinase domain (TKD).2 Impacting approximately 30 percent of AML patients,4 the FLT3-ITD mutation is associated with worsened disease free survival and overall survival.5,6 FLT3-TKD mutations impact approximately 7 percent of AML patients4 and, although the impact of these mutations is less clear,7 they have been associated with treatment resistance.8
“Our ability to use precision medicine to help patients with FLT3-mutated AML takes an important step forward with the approval of XOSPATA,” said Alexander Perl, M.D., Abramson Cancer Center, University of Pennsylvania. “There is an urgent need in the clinic for more targeted agents to help patients whose disease is either refractory to the initial therapy, or who have relapsed.”
“XOSPATA offers new hope to patients for whom the treatment path forward is unclear,” said Steven Benner, M.D., senior vice president and global therapeutic area head, Oncology Development, Astellas. “For the first time, people with relapsed or refractory FLT3 mutation-positive AML have an FDA approved FLT3-targeting treatment available to them. The approval of XOSPATA is also a proud, landmark moment for our oncology program and marks the first approval of a medicine that will be the cornerstone of our new presence in blood cancers.”
The FDA's approval of XOSPATA was based on the interim analysis of the following endpoints in the ADMIRAL clinical trial: the rate of complete remission (CR)/complete remission with partial hematologic recovery (CRh); the duration of CR/CRh (DOR); and the rate of conversion from transfusion dependence to transfusion independence. The CR/CRh rate was 21%. The median duration of CR/CRh was 4.6 months. The rate of conversion from transfusion dependence to transfusion independence was 31.1% for any 56 day post-baseline period. For patients who achieved a CR/CRh, the median time to first response was 3.6 months (range, 0.9 to 9.6 months). The CR/CRh rate was 29 of 126 in patients with FLT3-ITD or FLT3-ITD/TKD and 0 of 12 in patients with FLT3-TKD only.
Full results from the ADMIRAL trial will be submitted for presentation at an upcoming medical meeting.
The safety evaluation of XOSPATA was based on 292 patients with relapsed or refractory AML treated with 120 mg gilteritinib daily. The median duration of exposure to XOSPATA was 3 months (range 0.1 to 42.8 months). The most frequent non-hematological serious adverse reactions (≥5%) reported in patients were pneumonia (19%), sepsis (13%), fever (13%), dyspnea (7%) and renal impairment (5%). Overall, 22 of 292 patients (8%) discontinued XOSPATA treatment permanently due to an adverse reaction. The most common adverse reactions (>1%) leading to discontinuation were pneumonia (2%), sepsis (2%) and dyspnea (1%). The most common adverse reactions (≥20%) were myalgia/arthralgia (42%), transaminase increased (41%), fatigue/malaise (40%), fever (35%), non-infectious diarrhea (34%), dyspnea (34%), edema (34%), rash (30%), pneumonia (30%), nausea (27%), stomatitis (26%), cough (25%), headache (21%), hypotension (21%), dizziness (20%) and vomiting (20%).
Previously, XOSPATA was granted both Orphan Drug designation9 and Fast Track10 designation by the U.S. FDA. Gilteritinib also received Orphan Designation from the European Commission (EC)11 and Orphan Drug Designation from the Japan Ministry of Health, Labor and Welfare (MHLW).12 The MHLW also granted SAKIGAKE designation to gilteritinib for FLT3mut+ relapsed/refractory AML and approved the treatment for this population in September 2018.13
Astellas reflected the impact from this approval in its financial forecast of the current fiscal year ending March 31, 2019.
About XOSPATA
XOSPATA is indicated for the treatment of adult patients who have relapsed or refractory Acute Myeloid Leukemia (AML) with a FMS-like tyrosine kinase 3 (FLT3) mutation as detected by an FDA-approved test.2
XOSPATA was discovered through a research collaboration with Kotobuki Pharmaceutical Co., Ltd., and Astellas has exclusive global rights to develop, manufacture and potentially commercialize XOSPATA.
Important Safety Information
Contraindications
XOSPATA is contraindicated in patients with hypersensitivity to gilteritinib or any of the excipients. Anaphylactic reactions have been observed in clinical trials.
Warnings and Precautions
Posterior Reversible Encephalopathy Syndrome (PRES) There have been rare reports of PRES with symptoms including seizure and altered mental status with XOSPATA. Symptoms have resolved after discontinuation of XOSPATA. A diagnosis of PRES requires confirmation by brain imaging, preferably MRI. Discontinue XOSPATA in patients who develop PRES.
Prolonged QT Interval XOSPATA has been associated with prolonged cardiac ventricular repolarization (QT interval). Of the 292 patients treated with XOSPATA in the clinical trial, 1.4% were found to have a QTc interval greater than 500 msec and 7% of patients had an increase from baseline QTc greater than 60 msec. Perform electrocardiogram (ECG) prior to initiation of treatment with XOSPATA, on days 8 and 15 of cycle 1, and prior to the start of the next two subsequent cycles. Interrupt and reduce XOSPATA dosage in patients who have a QTcF >500 msec. Hypokalemia or hypomagnesemia may increase the QT prolongation risk. Correct hypokalemia or hypomagnesemia prior to and during XOSPATA administration.
Pancreatitis There have been rare reports of pancreatitis in patients receiving XOSPATA in clinical studies. Evaluate patients who develop signs and symptoms of pancreatitis. Interrupt and reduce the dose of XOSPATA in patients who develop pancreatitis.
Embryo-Fetal Toxicity Based on findings in animals and its mechanism of action, XOSPATA can cause embryo-fetal harm when administered to a pregnant woman. Advise females of reproductive potential to use effective contraception during treatment with XOSPATA and for at least 6 months after the last dose of XOSPATA. Advise males with female partners of reproductive potential to use effective contraception during treatment with XOSPATA and for at least 4 months after the last dose of XOSPATA. Pregnant women, patients becoming pregnant while receiving XOSPATA or male patients with pregnant female partners should be apprised of the potential risk to the fetus.
Adverse Reactions
The most frequent non-hematological serious adverse reactions (≥5%) reported in patients were pneumonia (19%), sepsis (13%), fever (13%), dyspnea (7%) and renal impairment (5%).
Overall, 22 of 292 patients (8%) discontinued XOSPATA treatment permanently due to an adverse reaction. The most common adverse reactions (>1%) leading to discontinuation were pneumonia (2%), sepsis (2%) and dyspnea (1%). The most common adverse reactions (≥20%) were myalgia/arthralgia (42%), transaminase increased (41%), fatigue/malaise (40%), fever (35%), non-infectious diarrhea (34%), dyspnea (34%), edema (34%), rash (30%), pneumonia (30%), nausea (27%), stomatitis (26%), cough (25%), headache (21%), hypotension (21%), dizziness (20%) and vomiting (20%).
Other clinically significant adverse reactions occurring in ≤10% of patients included: electrocardiogram QT prolonged (7%), cardiac failure (grouped terms) (4%), pericardial effusion (3%), pericarditis (2%), differentiation syndrome (1%), anaphylactic reaction (1%) and posterior reversible encephalopathy syndrome (1%).
Lab Abnormalities: The most common lab abnormalities (>20%) that were Grade ≥3 that occurred ≥10% were: hypophosphatemia (12%), alanine aminotransferase increased (12%), hyponatremia (12%), aspartate aminotransferase increased (10%).
Drug Interactions
Combined P-gp and Strong CYP3A Inducers: Concomitant use of XOSPATA with a combined P-gp and strong CYP3A inducer decreases XOSPATA exposure which may decrease XOSPATA efficacy. Avoid concomitant use of XOSPATA with combined P-gp and strong CYP3A inducers.
Strong CYP3A inhibitors: Concomitant use of XOSPATA with a strong CYP3A inhibitor increases XOSPATA exposure. Consider alternative therapies that are not strong CYP3A inhibitors. If the concomitant use of these inhibitors is considered essential for the care of the patient, monitor patient more frequently for XOSPATA adverse reactions. Interrupt and reduce XOSPATA dosage in patients with serious or life-threatening toxicity.
Drugs that Target 5HT2B Receptor or Sigma Nonspecific Receptor: Concomitant use of XOSPATA may reduce the effects of drugs that target the 5HT2B receptor or the sigma nonspecific receptor (e.g., escitalopram, fluoxetine, sertraline). Avoid concomitant use of these drugs with XOSPATA unless their use is considered essential for the care of the patient.
Specific Populations
Lactation: Advise women not to breastfeed during treatment with XOSPATA and for 2 months after the last dose.
Please see Full Prescribing Information for additional safety information.
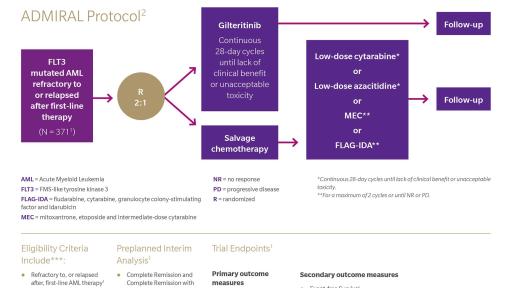
About the ADMIRAL Trial14
The Phase 3 ADMIRAL trial (NCT02421939) was an open-label, multicenter, randomized study of gilteritinib versus salvage chemotherapy in adult patients with FLT3 mutations who are refractory to or have relapsed after first-line AML therapy. The primary endpoints of the trial are Overall Survival (OS) and complete remission/complete remission with partial hematologic recovery (CR/CRh) rates. The study enrolled 371 patients with FLT3 mutations present in bone marrow or whole blood, as determined by central lab. Subjects were randomized in a 2:1 ratio to receive gilteritinib (120 mg15) or salvage chemotherapy.
About Astellas
Astellas Pharma Inc., based in Tokyo, Japan, is a company dedicated to improving the health of people around the world through the provision of innovative and reliable pharmaceutical products. For more information, please visit our website at www.astellas.com/us.
Cautionary Notes
In this press release, statements made with respect to current plans, estimates, strategies and beliefs and other statements that are not historical facts are forward-looking statements about the future performance of Astellas. These statements are based on management’s current assumptions and beliefs in light of the information currently available to it and involve known and unknown risks and uncertainties. A number of factors could cause actual results to differ materially from those discussed in the forward-looking statements. Such factors include, but are not limited to: (i) changes in general economic conditions and in laws and regulations, relating to pharmaceutical markets, (ii) currency exchange rate fluctuations, (iii) delays in new product launches, (iv) the inability of Astellas to market existing and new products effectively, (v) the inability of Astellas to continue to effectively research and develop products accepted by customers in highly competitive markets, and (vi) infringements of Astellas’ intellectual property rights by third parties.
Contacts for inquiries or additional information:
Astellas Pharma Inc.
Corporate Communications
TEL: +81-3-3244-3201 FAX: +81-3-5201-7473
United States:
Media inquiries: Marjorie Moeling
TEL: +1-224-205-5205 E-MAIL: [email protected]
1American Cancer Society. What is acute myeloid leukemia? (02-22-2016). https://www.cancer.org/cancer/acute-myeloid-leukemia/about/what-is-aml.html. Accessed 05-10-2018.
2XOSPATA [package insert]. Northbrook, IL: Astellas Inc.
3American Cancer Society. Key statistics for acute myeloid leukemia (01-04-2018). https://www.cancer.org/cancer/acute-myeloid-leukemia/about/key-statistics.html. Accessed 03-12-2018.
4Patel JP, Gönen M, Figueroa ME, et al. Prognostic relevance of integrated genetic profiling in acute myeloid leukemia. N Engl J Med. 2012;366(12):1079-89.
5Whitman SP, Archer KJ, Feng L, et al. Absence of the wild-type allele predicts poor prognosis in adult de novo acute myeloid leukemia with normal cytogenetics and the internal tandem duplication of FLT3: a Cancer and Leukemia Group B study. Cancer Res. 2001;61(19):7233-7239.
6Whitman SP, Maharry K, Radmacher MD, et al. FLT3 internal tandem duplication associates with adverse outcome and gene- and microRNA-expression signatures in patients 60 years of age or older with primary cytogenetically normal acute myeloid leukemia: a Cancer and Leukemia Group B study. Blood. 2010;116(18):3622-3626.
7Bacher U, Haferlach C, Kern W, Haferlach T, Schnittger S. Prognostic relevance of FLT3-TKD mutations in AML: the combination matters—an analysis of 3082 patients. Blood. 2008;111(5):2527-37.
8Alvarado Y, Kantarjian H, Luthra R, et al. Treatment with FLT3 inhibitor in patients with FLT3-mutated acute myeloid leukemia is associated with development of secondary FLT3–tyrosine kinase domain mutations. Cancer. 2014;120(14):2142-9.
9Astellas Pharma Inc. U.S. FDA Grants Orphan-Drug Designation to Astellas for Development of FLT3 Inhibitor Gilteritinib in Acute Myeloid Leukemia (07-20-2017). https://newsroom.astellas.us/2017-07-20-U-S-FDA-Grants-Orphan-Drug-Designation-to-Astellas-for-Development-of-FLT3-Inhibitor-Gilteritinib-in-Acute-Myeloid-Leukemia. Accessed 11-21-2018.
10Astellas Pharma Inc. U.S. FDA Grants Priority Review to Astellas' New Drug Application for Gilteritinib for the Treatment of Adult Patients with Relapsed or Refractory Acute Myeloid Leukemia (AML) (05-29-2018). https://www.astellas.com/us/news/806. Accessed 11-21-2018.
11Astellas Pharma Inc. Astellas Receives Orphan Designation from the European Commission for Gilteritinib for the Treatment of Acute Myeloid Leukaemia (07-23-2018). https://www.astellas.com/en/news/10211. Accessed 11-21-2018.
12Astellas Pharma Inc. Astellas Receives Orphan Drug Designation from the Japanese MHLW for Gilteritinib (03-22-2018). https://www.astellas.com/en/news/10466. Accessed 11-21-2018.
13Astellas Pharma Inc. Astellas Announces Approval in Japan for XOSPATA® 40 mg Tablets for the Treatment of FLT3mut+ Relapsed or Refractory AML (09-21-2018). https://www.astellas.com/us/news/14271. Accessed 11-21-2018.
14ClinicalTrials.gov. A study of ASP2215 versus salvage chemotherapy in patients with relapsed refractory acute myeloid leukemia (AML) with FMS-like tyrosine kinase (FLT3) mutation (04-25-2018). https://clinicaltrials.gov/ct2/show/NCT02421939. Accessed 04-26-2018.
15Gorcea CM, Burthem J, Tholouli E. ASP2215 in the treatment of relapsed/refractory acute myeloid leukemia with FLT3 mutation: background and design of the ADMIRAL trial. Future Oncol (Epub) 03-02-2018.








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}